\(\alpha\)-Fe with ferromagnetic calculation¶
In this example, you will learn how to perform a ferromagnetic DFT+G calculation using Wien2k plus CyGutz packages, including some typical post-analyses.
- Finish a self-consistent LDA paramagnetic calculation (run_lapw)
for \(\alpha\)-Fe (bcc) using Wien2k. Here is the structure file
Fe.struct. To compare with the provided results, one should keep the RMT = 2.33 as specified in the Fe.struct, RMT * KMAX = 8.0 and total number of k-points = 5000 (17x17x17). We do not shift k-points here. In the end, check the total LDA energy in the Fe.scf file, which should be close to -2541.12378 Ryd.
- Use the following command to initialize the Gutzwiller calculation,
Type:
$ ${WIEN_GUTZ_ROOT2}/init_ga.pyAnswer the questions as follows:
- Do you want to BREAK SPIN-SYMMETRY: y
- Do you want to COMPLETELY break orbital-symmetry: n
- Do you want to take into account the SPIN-ORBIT interaction: n
- Do you want to take into account the CRYSTAL FIELD effect: y
- Please select the method to parametrize Coulomb U-matrix: 1
- Please select method for U-interaction double counting: 12
- Symmetrically-equivalent atom indices …: y
- Enter up(1) dn(-1) or 0 for spin-moment of the atoms: 1
- Is this atom correlated: y
- Enter correlated shells: d
- Please provide interaction parameters U,J: 7.0 0.8
- Please provide initial guess … localized d-electrons: 6.5
- Please select the method to solve G-RISB equations: 0
- Please select the method to solve embedding Hamiltonian: -1
Check the file init_ga.slog and you will see that the local self-energy structure has the following form in the single-particle basis with spin (up,down) as the faster index:
[[1 0 0 0 0 0 0 0 0 0] [0 3 0 0 0 0 0 0 0 0] [0 0 1 0 0 0 0 0 0 0] [0 0 0 3 0 0 0 0 0 0] [0 0 0 0 1 0 0 0 0 0] [0 0 0 0 0 3 0 0 0 0] [0 0 0 0 0 0 2 0 0 0] [0 0 0 0 0 0 0 4 0 0] [0 0 0 0 0 0 0 0 2 0] [0 0 0 0 0 0 0 0 0 4]]
Different integers are used for the spin-up and down conponents, inidcating spin-polarization. The self-energy is diagonal due to cubic symmetry.
To set up the magnetic configuration, type:
$ ${WIEN_GUTZ_ROOT2}/init_magnetism.pyAnswer the questions as follows:
- enter spin up or down: up
- please enter the magnitude of the field: 0.3
- Is the external field applied only at initial step (0) …: 0
Here we add a 0.3 eV/Bohr magneton local magnetic field to break spin symmetry INITIALLY.
- Type the command below to
run the DFT+G calculation:
$ ${WIEN_GUTZ_ROOT2}/run_ga.pyAfter convergence, check the total energy in Fe.scf file, which should be close to -2540.94918 Ryd. You can also find
total magnetic moment= 2.14 in the main output text file GUTZ.LOG.
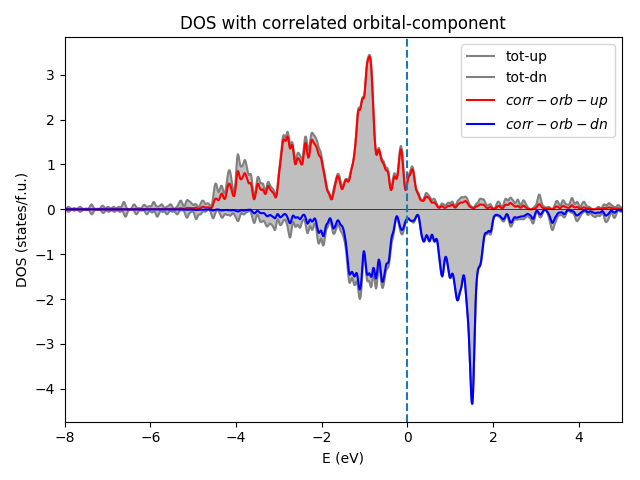
- To plot the spin-resolved density of states
with overall Fe-3d character, type:
$ ${WIEN_GUTZ_ROOT2}/plot_dos_tf.pyyou will get figure as below
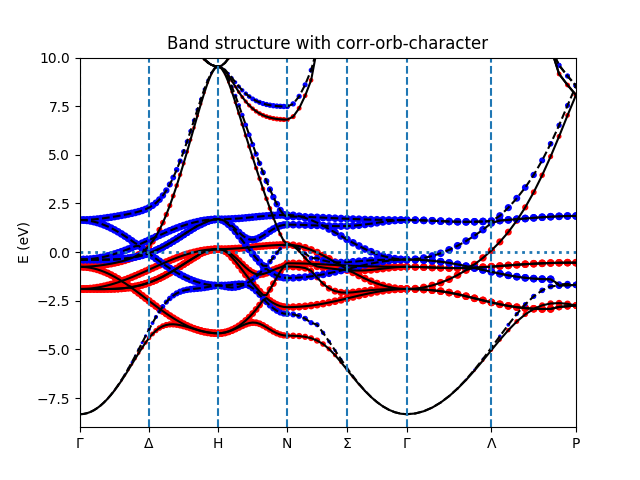
- To calculate the bands structure along selected k-path,
follow the steps below:
Prepare the fe.klist_band file for the high-symmetry k-path of the primitive Brillouin Zone. The SRC_templates directory of Wien2k has some examples. For instance, we can use bcc.klist file. Type the command to get the file:
$ cp ${WIENROOT}/SRC_templates/bcc.klist Fe.klist_bandType the following command to calculate the band structure:
$ ${WIEN_GUTZ_ROOT2}/run_ga.py -bandTo plot spin-resolved band structure with Fe-3d character, type:
$ ${WIEN_GUTZ_ROOT2}/plot_band_tf.py -h # help info $ ${WIEN_GUTZ_ROOT2}/plot_band_tf.py -el -8 -eh 10You will see the band structure like the following